
A tool for the optimal design of sgRNAs for simultaneous cleavage of multiple genes.
Given a set of genomic sequences as input, CRISPys designs the most promising sgRNAs to target all with high efficiency.
The webserver proposes four design strategies:
- A single sgRNA that could best target the entire gene set
This strategy can be executed by selecting the "Consider all genes" optimization criterion. (default) - A single sgRNA optimized to target most of the genes, each gene with a score above a given threshold Ω
This strategy can be executed by selecting the "Consider only genes above a threshold" optimization criterion. - Multiple sgRNAs, each directed towards subgroup of homologous genes
This strategy can be executed by confirming the "Consider gene homology" button. - The minimal set of sgRNAs that could target the entire gene set with high efficiency
Automatically computed for each execution of strategy II.
Usage options
The scoring function
specifies the estimated targeting efficiency of a given sgRNA to a given genomic site. The site currently supports three scoring functions:
- The CFD score [1]
- Optimized CRISPR Design [2]
- CCTop [3]
The CCTop score is divided by -224 (the minimal potential score) to provide a score between [0,1].
Additional scoring functions may be integrated upon request.
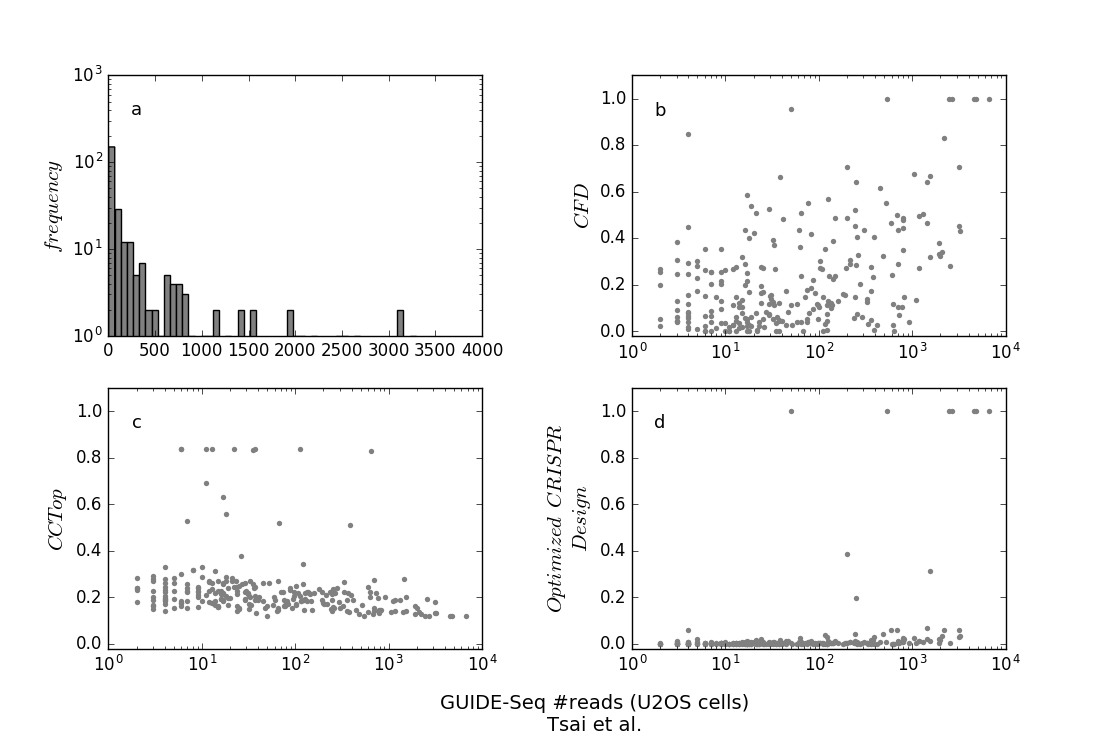
(a) The frequencies of the binned #reads. (b-d) The computed scores by the CFD score [1], CCTop [2], and Optimized CRISPR Design [3].
Optimization criterion
CRISPys proposes two criteria for the optimization of sgRNA design:
- The sum of scores across the input sequences (calculated by the selected scoring function).
- The number of sequences that could be cleaved with a score greater than a chosen threshold value. Setting this threshold to 0.0 will result in all input genes, while setting it to 1.0 will practically search for the sgRNA that perfectly matches the largest number of genes. We recommend the thresholds of 0.45, which represents the average CFD score of the targets between the 90th and 95th percentiles in the data by Tsai et al. [4] (see above).
Consider gene homology
Enabling this option would execute design strategy III. For selection of proper thresholds, please see the distribution of the scoring functions presented above.
Dividing the input genes set into smaller groups according to sequence similarity would increase the flexibility of the screen and allow a more focused experimental design. To this end, this strategy recursively splits the input genes into homologous subgroups and generates potential sgRNAs for each of them.
Practically, up to 10 sgRNAs will be designed for each subgroup of size two to nine.
Results Page:
The results for your run are denoted in a table, sorted by the optimal score of the sgRNAs.
Columns:
- sgRNA: the potential sgRNA designed by CRISPys.
- score: sum of cleavage scores for the genes targeted by the designed sgRNA (Genes scores).
- Genes, Genes scores, Target site: list of genes targeted by the sgRNA, with their cleavage score (calculated by the chosen scoring function), and the target site in the sequence.
Lower-case letters represent mismatches to the sgRNA. The first and default row holds the gene with highest cleavage score. Press the "+" sign to see the full list of genes
When the "Consider gene homology" is enabled, every section of the table corresponds to a homologous genes subgroup as specified by the internal nodes of the constructed genes tree. The name of the subgroup and the list of genes are given in the header of each section.
Often multiple sgRNAs are designed for a single subset of targets. The website presents only the best sgRNA for every subset of targets by default. You may see all possible designs by unfiltering the table.
References:
[1] Doench, John G., et al. "Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9." Nature biotechnology 34.2 (2016): 184.
[2] Zhang Lab, Optimized CRISPR Design, Mit. (2013) 2013. http://crispr.mit.edu/.
[3] M. Stemmer, T. Thumberger, M. del Sol Keyer, J. Wittbrodt, J.L. Mateo, CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool, PLoS One. 10 (2015) e0124633. doi:10.1371/journal.pone.0124633.
[4] Tsai, Shengdar Q., et al. "GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases." Nature biotechnology 33.2 (2015): 187-197.